
目次
1. pysamとSAMファイルの概要
1.1 pysamとは
1.2 SAM/BAMファイル
1.3 ヘッダー
1.4 アライメント
2. pysamの実験
2.1 環境構築
2.2 データロード
2.3 データ表示
1. pysamとは
1.1 Pysam
Pysam は、htslib C-API の軽量ならラッパーです。BAM/SAMファイルなどのDNAの塩基配列の読み取り、操作、および書き込みPython モジュールができます。対応ファイルはSAM/BAM/VCF/BCF/BED/GFF/GTF/FASTA/FASTQです。SamtoolsとBcftoolsの関数を利用できます。
Github: https://github.com/pysam-developers/pysam
資料:https://pysam.readthedocs.io/en/latest/
1.2 SAM/BAMファイル
SAMはSequence Alignment Mapの略称で、アライメントしたリードの情報を格納するテキスト形式です。
オプションのヘッダー セクションとアライメントセクションで構成されるタブ区切りのテキスト形式です。
BAMはバイナリ形式のSAMファイルです。
CRAMはSAM/BAM高圧縮アライメントファイルです。
ファイル拡張子: .sam、.bam、.cram (.gz、.bgz、.bz2、.xz)
SAM ファイルの説明資料:https://samtools.github.io/hts-specs/SAMv1.pdf
ヘッダーとアライメントの例
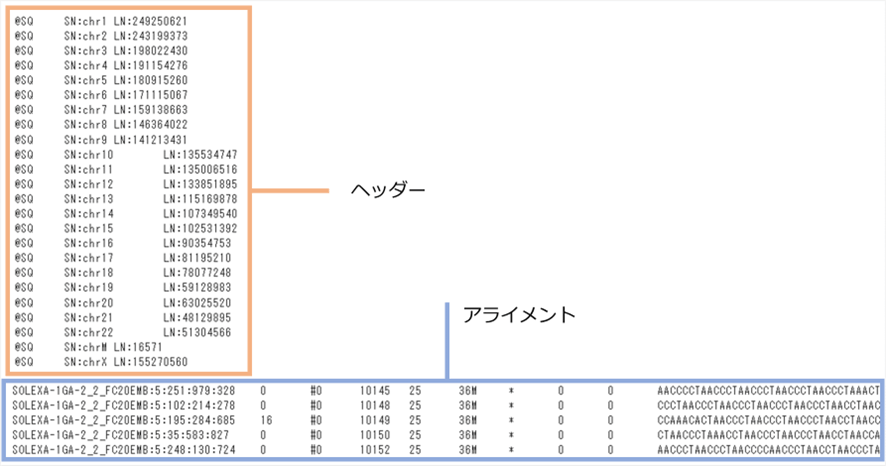
1.3 ヘッダー
各ヘッダー行は@から始まる二文字のコードが記載されています。タグのコードは下記になります。
@HD ファイルヘッダー
VN SAMフォーマットのバージョン
SO アライメントのソート情報。「unknown」、「unsorted」、「queryname」または「coordinate」のいずれか記載される
GO アライメントのグループ
SS アラインメントのサブソート順
@SQ リファレンス配列の情報
SN リファレンス配列の名前
LN リファレンス配列の長さ
AH 配列が代替遺伝子座であること
AN 代替リファレンス配列名
AS ゲノムアセンブリーID
DS 説明
M5 MD5 checksum
SP 生物種
TP 種族
UR 配列のURI
@RG リードのグループ
ID リードのグループのID
BC バーコード
CN リードを生成するシーケンシング センターの名前
DS 説明
DT 実行の日付
FO フロー順序
KS ヌクレオチド塩基の配列
LB ライブラリ
PG プログラム
PI 予測されるインサート サイズの中央値
PL プラットホーム
PM プラットフォーム モデル
PU プラットフォームユニット
SM サンプル
@PG プログラム
ID マッピングを行ったプログラム
PN プログラム名
CL マッピングを行うときに実際に入力したコマンドが記載される
PP 前の @PG-ID
DS 説明
VN プログラムのバージョン
1.4 アライメント
各アライメント行11列からなるタブ区切りのフィールドであらわされます。
- QNAME リードの名前。情報がない場合、’*’となります。
- FLAG 下記のマッピングの状態を示すビット値です。
・ 0 フォワード ストランドにマッピングされます
・ 1 0x1 ペアリードがある
・ 2 0x2 両方マップされている
・ 4 0x4 read1 はマップされていない
・ 8 0x8 read2(相方) はマップされていない
・ 16 0x10 read1 は逆相補鎖にマップされている
・ 32 0x20 read2(相方) は逆相補鎖にマップされてる
・ 64 0x40 テンプレートの最初のセグメント(read1)
・ 128 0x80 テンプレートの最後のセグメント(read2)
・ 256 0x100 複数個所にマップされている
・ 512 0x200 プラットフォーム/ベンダーの品質管理などのフィルターを通過しない
・ 1024 0x400 PCR または光学的複製
・ 2048 0x800 補足アライメント
- RNAME リファレンス配列の名前。マッピングされなかったリードは * となります。
- POS(POSition)マッピングされたポジション。1始まり。マッピングされなかったリードは0となります。
POSの例:
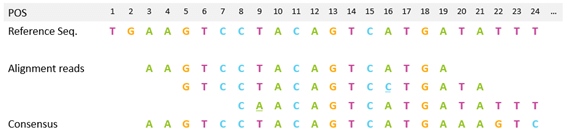
- MAPQ(MAPping Quality)マッピングポジションが誤っている確率の常用対数に-10を掛けた値です。255はマッピングクオリティが得られなかったことを示す。
- CIGAR (Compact Idiosyncratic Gapped Alignment Report)リファレンスに対し、どのようにアライメントされたかを示します。
M 参照配列に張り付いたリード。ただし、個々の塩基が参照配列と同じかどうかは感知しない。
I 挿入
D 欠失
N 間が飛んでいる。mRNAではイントロンがメイン
S ソフトクリップ。張り付いていない端っこのリード。何が張り付いていないのかは明記されている。
H ハードクリップ。張り付いていない端っこのリード。何が張り付いていないのかは不明。
P padding。張り付いたリードの側で何かの欠失が起こっている。de novoがメインしているか。
= 参照配列に張り付いたリードのうち、参照配列と同じ塩基。
X 参照配列に張り付いたリードのうち、参照配列とは違う塩基。
CIGARの例:

- MNAME paired-endの場合、もう一方のリードの名前を示す。”=”の場合、もう一方のリードも同じ名前であることを示します。
- MPOS 最もよくマッピングされたポジションを示します。
- TLEN 全てのセグメントが同一のリファレンス配列にマッピングされた場合、最も左側にマップされた塩基から最も右側にマップされた塩基までの長さが計算される (end − start + 1)。マップされたか否かはCIGARフォーマットから判別される(したがってsoft-clipped部分は含まれない)。最も左のセグメントは正の符号が、最も右のセグメントは負の符号がつく。この列は、対応するリードがどこにアラインされているかを把握するために存在する。コンセンサスの取れた定義は存在せず、ファイルの出力に使用したツールなどに依存しています。
- SEQ リファレンスとして使用されるリード中の配列。”*” でない場合、CIGAR列のM/I/S/=/Xを足したものになります。
- QUAL リードのクオリティー。fasta/q format中のものと同じ(Phredクオリティスコア + 33 をASCIIコードにエンコードした値)
2. pysamの実験
Google Colabの環境で実験します。
データセット:人の24この染色体のBAMファイル
詳細:http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwRepliSeq/
2.1 環境構築
ライブラリのインストール
| !pip install -q pysam |
ライブラリのインポート
| import pysam pysam.__version__ |
0.19.1
2.2 データロード
UcscからBG02ES.bamのファイルをダウンロードします。
BG02ESはヒト胚性幹細胞のデータです。
| import urllib from urllib import request
src_file = “http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeUwRepliSeq/wgEncodeUwRepliSeqBg02esG1bAlnRep1.bam” dest_file = ‘BG02ES.bam’
urllib.request.urlretrieve(src_file, dest_file) |
ファイルの読み込み
| file_path = “/content/BG02ES.bam” samfile = pysam.AlignmentFile(file_path, “rb”) |
2.3 データ表示
ヘッダーを表示します。下記のようなデータを表示します。
SQ リファレンス配列辞書
SN リファレンス配列名 24この染色体
LN リファレンス配列の長さ
| print(samfile.header) |
@SQ SN:chr1 LN:249250621
@SQ SN:chr2 LN:243199373
@SQ SN:chr3 LN:198022430
@SQ SN:chr4 LN:191154276
@SQ SN:chr5 LN:180915260
@SQ SN:chr6 LN:171115067
@SQ SN:chr7 LN:159138663
@SQ SN:chr8 LN:146364022
@SQ SN:chr9 LN:141213431
@SQ SN:chr10 LN:135534747
@SQ SN:chr11 LN:135006516
@SQ SN:chr12 LN:133851895
@SQ SN:chr13 LN:115169878
@SQ SN:chr14 LN:107349540
@SQ SN:chr15 LN:102531392
@SQ SN:chr16 LN:90354753
@SQ SN:chr17 LN:81195210
@SQ SN:chr18 LN:78077248
@SQ SN:chr19 LN:59128983
@SQ SN:chr20 LN:63025520
@SQ SN:chr21 LN:48129895
@SQ SN:chr22 LN:51304566
@SQ SN:chrM LN:16571
@SQ SN:chrX LN:155270560
リファレンス配列名を表示します。
| print(samfile.references) print(samfile.nreferences) |
(‘chr1’, ‘chr2’, ‘chr3’, ‘chr4’, ‘chr5’, ‘chr6’, ‘chr7’, ‘chr8’, ‘chr9’, ‘chr10’, ‘chr11’, ‘chr12’, ‘chr13’, ‘chr14’, ‘chr15’, ‘chr16’, ‘chr17’, ‘chr18’, ‘chr19’, ‘chr20’, ‘chr21’, ‘chr22’, ‘chrM’, ‘chrX’)
24
リファレンス配列の長さを表示します。
| print(samfile.lengths) |
(249250621, 243199373, 198022430, 191154276, 180915260, 171115067, 159138663, 146364022, 141213431, 135534747, 135006516, 133851895, 115169878, 107349540, 102531392, 90354753, 81195210, 78077248, 59128983, 63025520, 48129895, 51304566, 16571, 155270560)
インデックスを作成します。
| # idex作成 file_path = “/content/BG02ES.bam” pysam.index(file_path) samfile = pysam.AlignmentFile(file_path, “rb”) |
特定のリファレンス配列の長さを表示します。
| samfile.count(“chr1”) |
179687
アライメントを表示します。
| samfile = pysam.AlignmentFile(file_path, “rb”) for idx, read in enumerate(samfile.fetch(reference=’chr1′)): if idx = 1: print(read) samfile.close() |
SOLEXA-1GA-2_2_FC20EMB:5:251:979:328 0 #0 10145 25 36M * 0 0 AACCCCTAACCCTAACCCTAACCCTAACCCTAAACT array(‘B’, [71, 71, 71, 71, 39, 66, 54, 71, 71, 39, 51, 70, 71, 66, 42, 32, 62, 46, 45, 71, 32, 32, 36, 36, 33, 57, 36, 30, 39, 30, 34, 33, 34, 30, 35, 32]) [(‘NM’, 1), (‘X1’, 1), (‘MD’, ’33A2′)]

担当者:KW
バンコクのタイ出身 データサイエンティスト
製造、マーケティング、財務、AI研究などの様々な業界にPSI生産管理、在庫予測・最適化分析、顧客ロイヤルティ分析、センチメント分析、SaaS、PaaS、IaaS、AI at the Edge の環境構築などのスペシャリスト